Today went really great, I was able to conduct a western blot using gelatin and an electrical supply. Western blots tell you the different levels of proteins in different parts of the cells nucleus.
I entered the lab and I was going to do a gel run. Essentially, you have these slides and you put nitrocellulose that has been washed with rabbit and goat protein onto the slide. Nitrocellulose is a material that is actually made from pure cellulose. Pure cellulose is what makes up cotton. It can be put into a paper-like form by a process in which the nitrocellulose is dissolved in a solvent. Nitrocellulose was what used to be used to make movie film and various plastics. Although it was of great use, it was soon found that it was far too flammable for practical use. I have made it many times, and on ignition it always flashes and makes a satisfying “whoompf” noise that never ceases to bring a smile to my face. The proteins are washed onto the slide and what is interesting is that these proteins actually help the cell samples to cling to the nitrocellulose and stain it. Think of it sort of like a chain link. You have rabbit protein which clings to goat proteins, and then the goat proteins cling to the cancer cell proteins that are in the nucleus. There is this slide, and it has these little wells, you fill the wells with your cancer cell samples. The slide is filled with electro-conductive gelatin. The protein samples stain and make these bands at the different points depending on the weight of the protein. You also have what is called a molecular weight. The molecular weight is essentially a control. The molecular weight always falls onto one band. This way depending on the density of the band, you can tell how much of a certain protein was active and formulated inside the cell, this shows in better detail how the cell reacted to the drug and antibody. I will post some pictures in the next week’s post of the western blot results as it is hard to explain in just words. 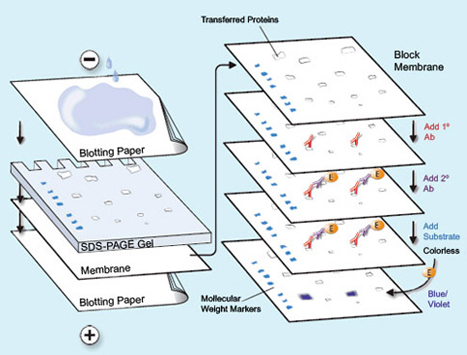
Again, comments are very welcome and I hope you enjoyed.
Thank you for your time,
Aaron J.



